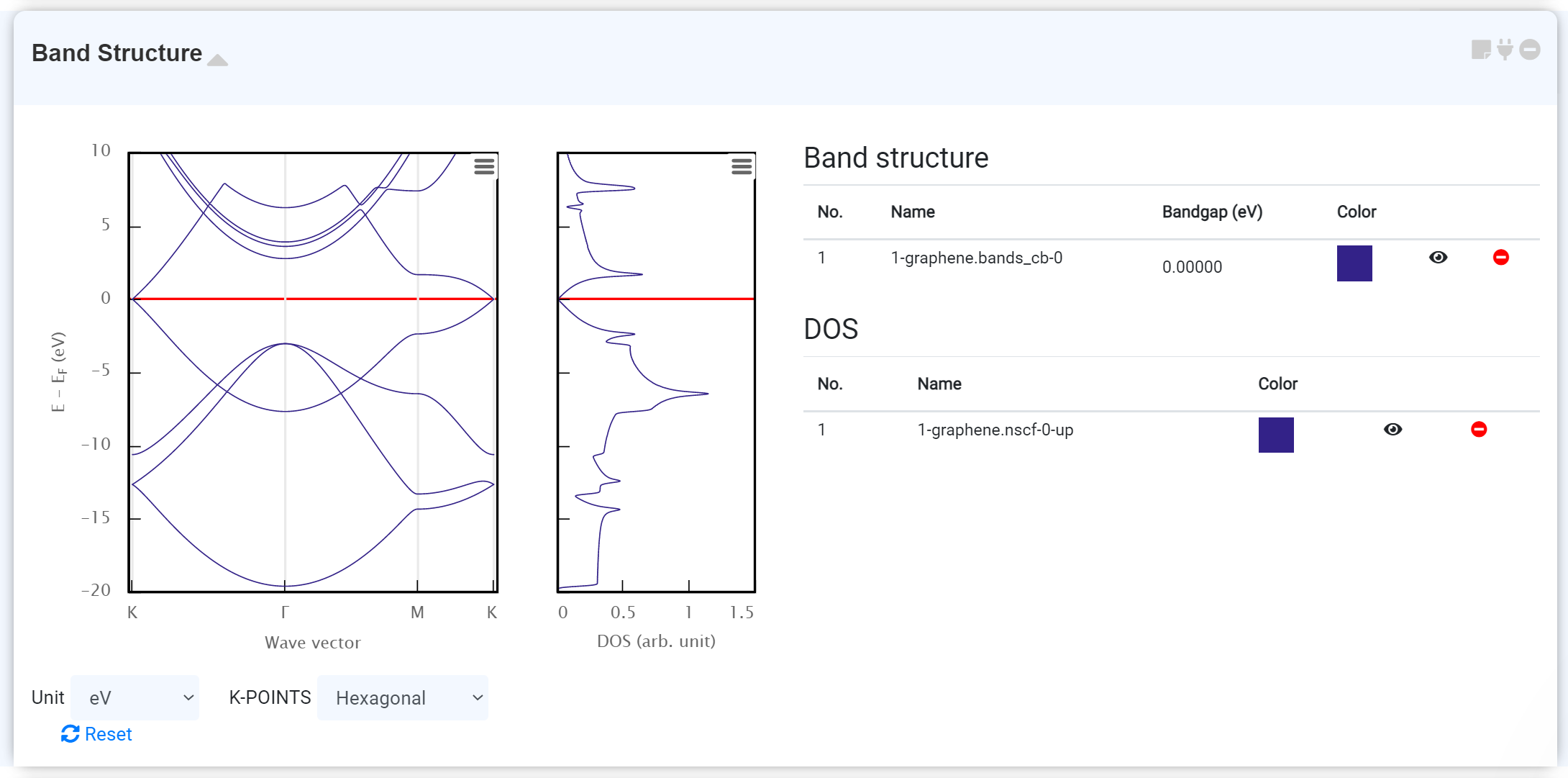
Band Structure
You can get the band structure of materials, which can be used to characterize the band gap of the system.
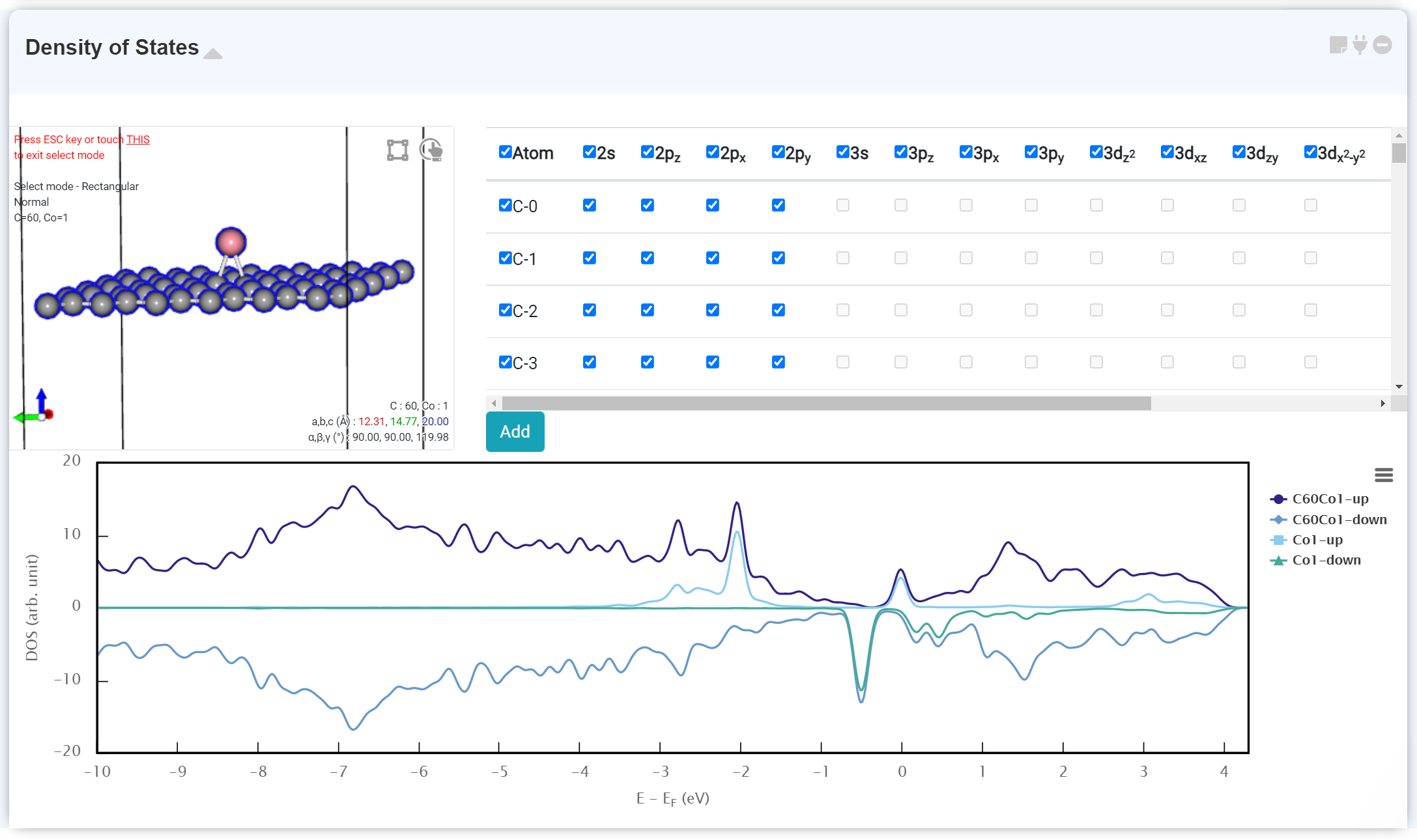
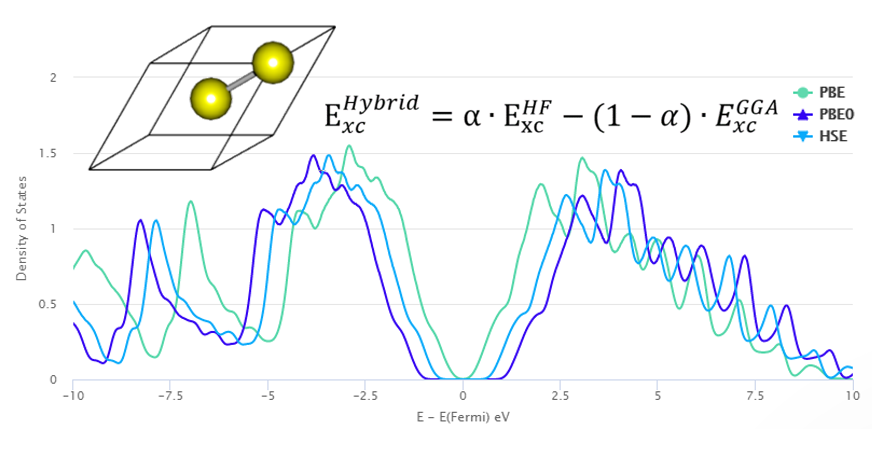
Density of States
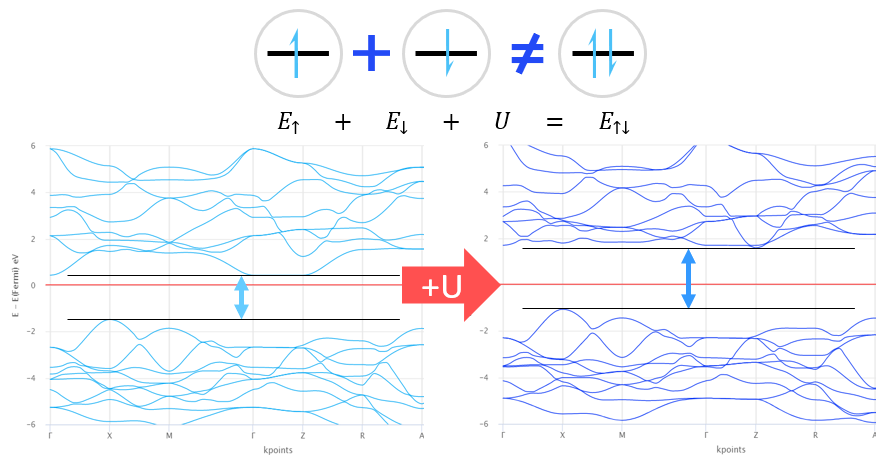
Atom and orbital-projected density of states and the entire density of states of the system can be analyzed. This analysis can be used for example investigating the effect of the defect on the electron structure of the magnetic properties of the system.
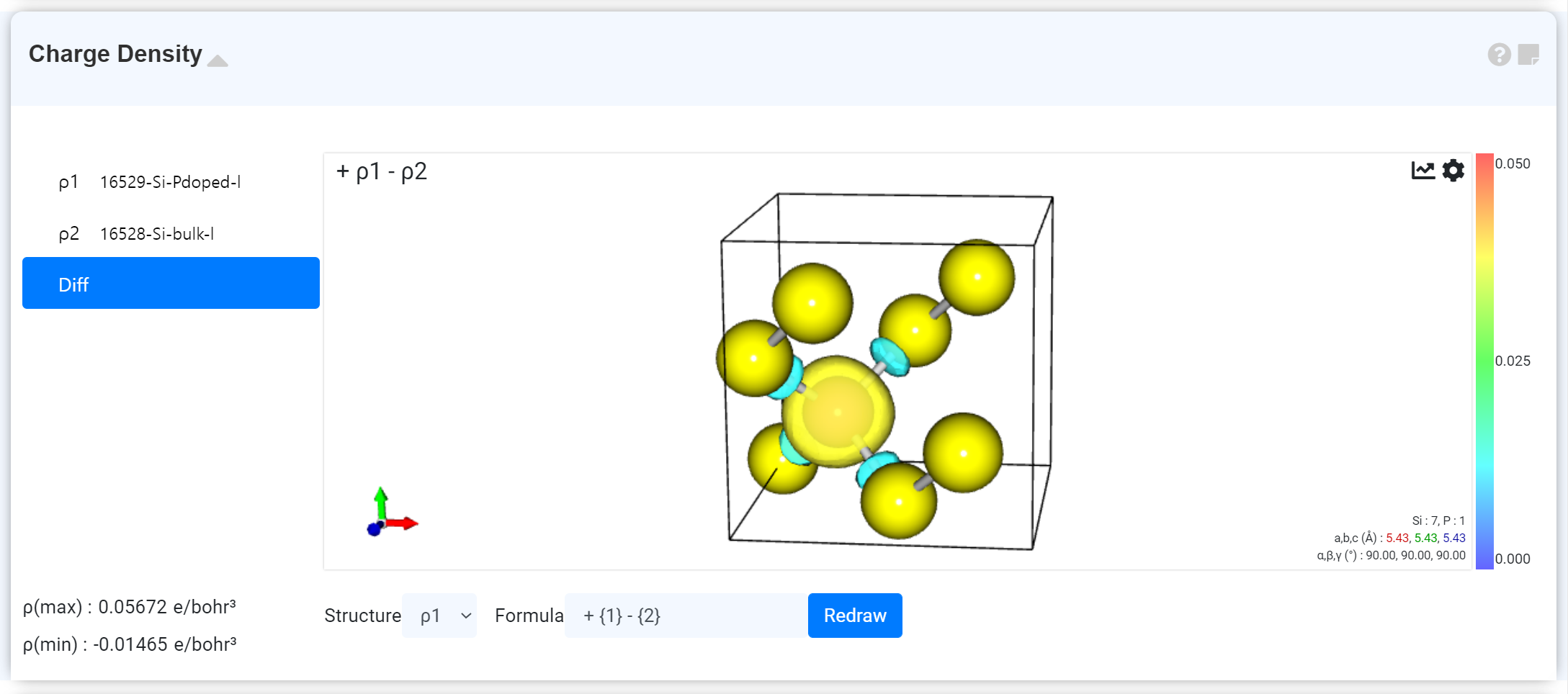
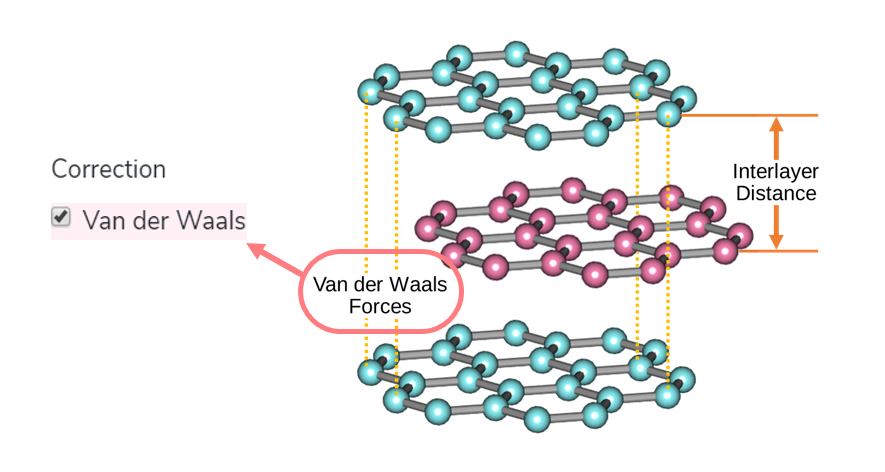
Charge Density
You can conveniently find and visualize the charge density data by carrying out a plane-wave basis DFT calculation of Quantum Espresso.
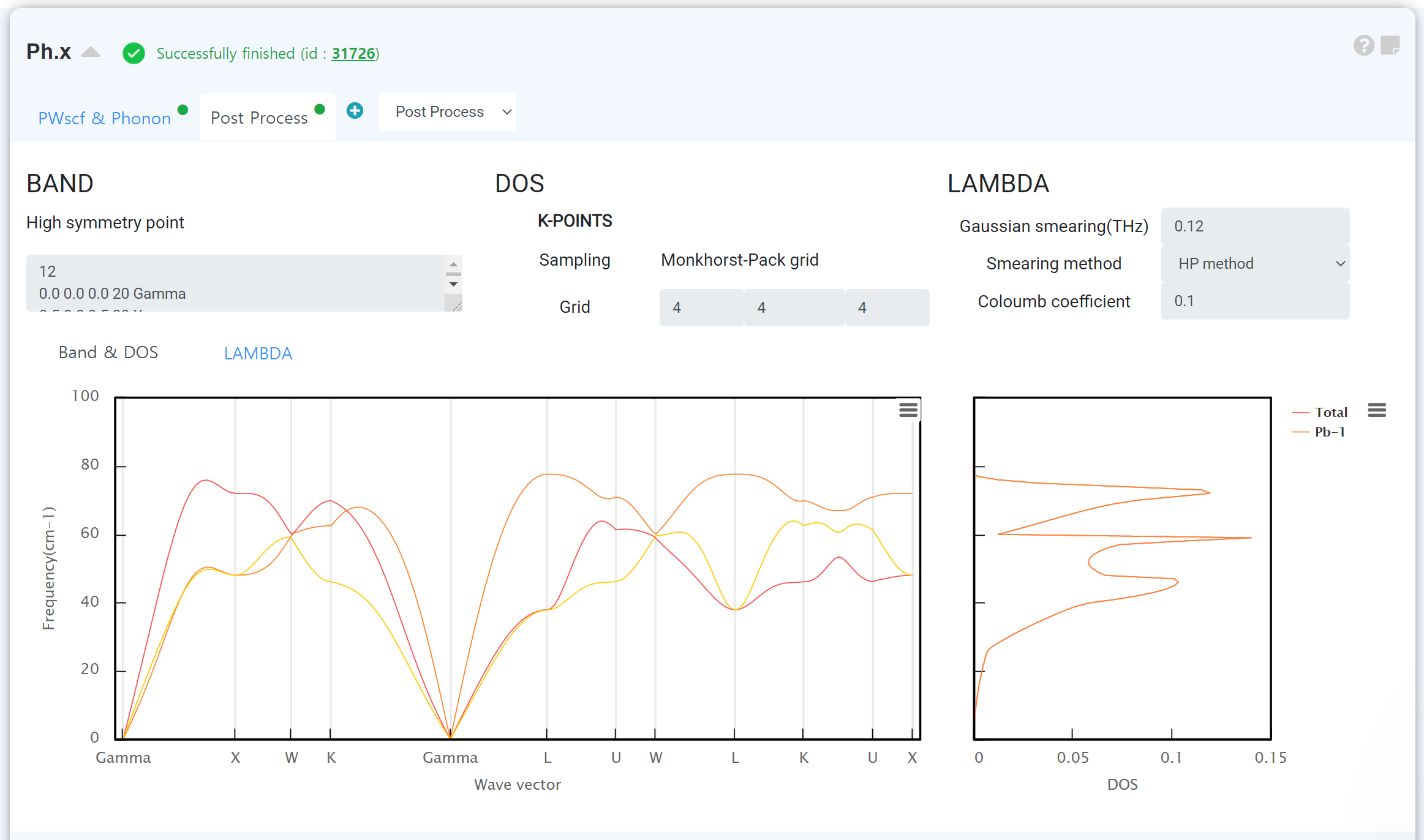
Phonon
You can get the phonon dispersion and other phonon-related properties of materials.
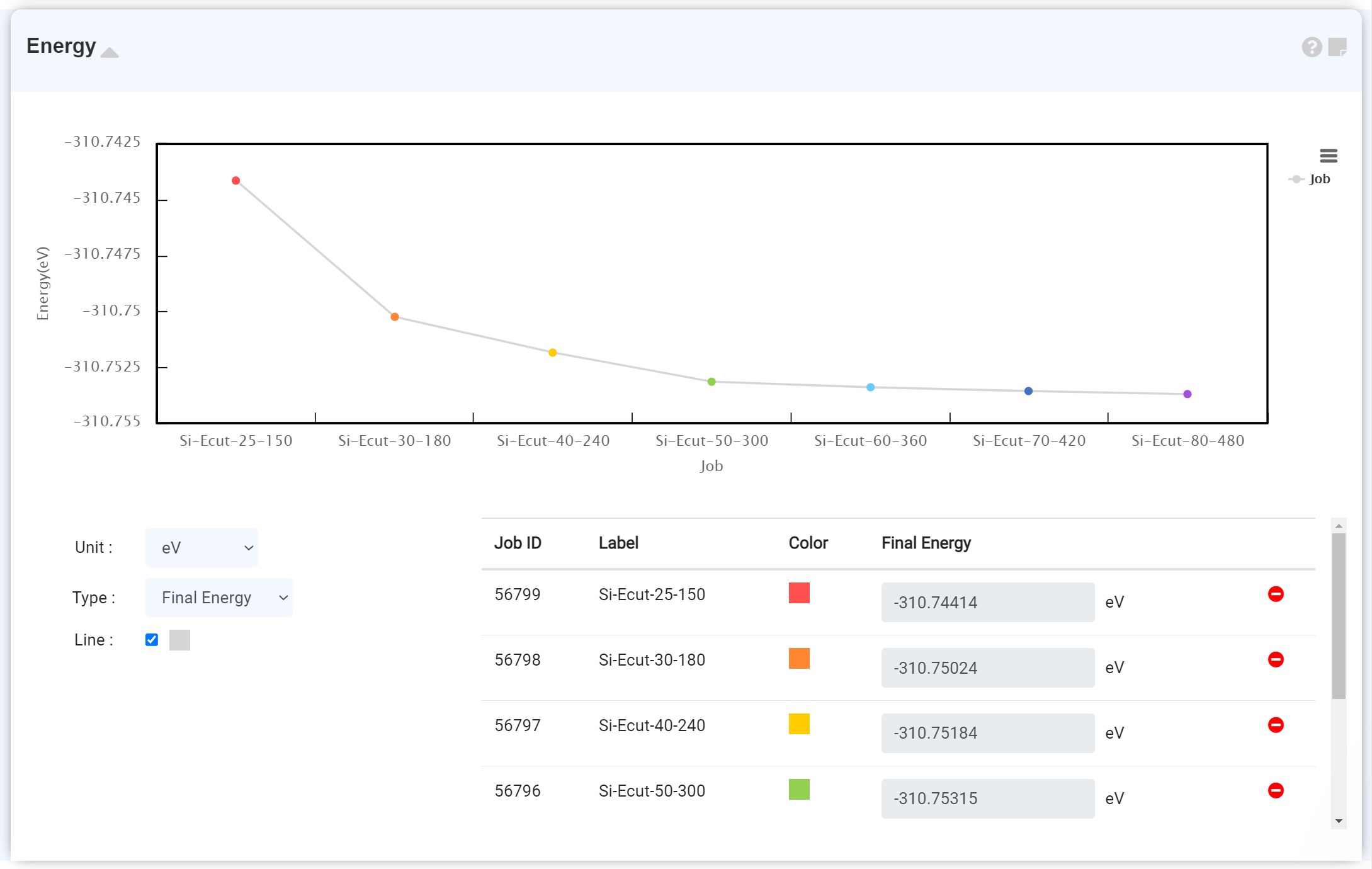
Energy Comparison
The energies of different calculations can be visually compared. This comparison will enable you to friendly the most probable adsorption site on surface or the equilibrium dopant position.
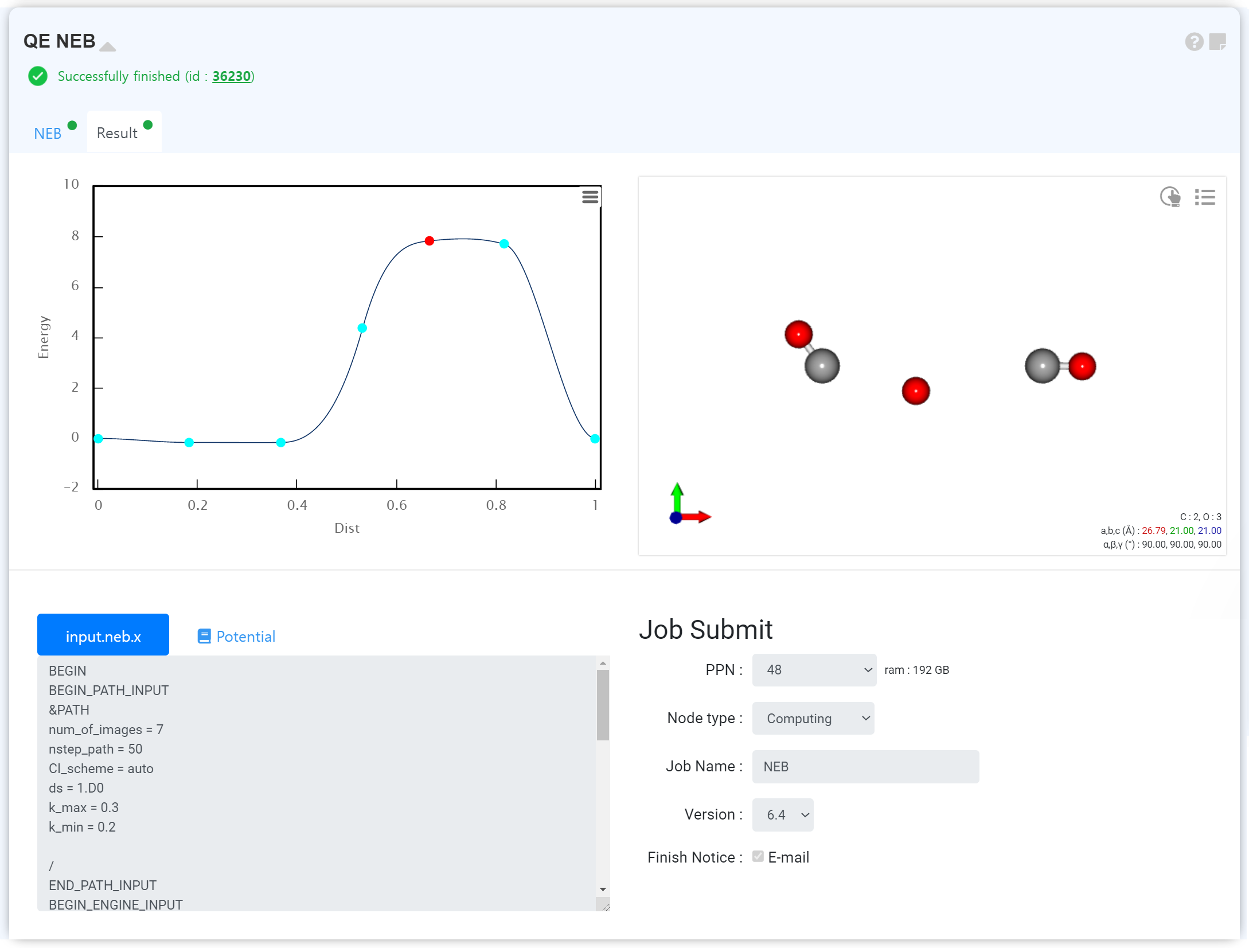
NEB Calculation
You can analyze the diffusion of atoms and dopants, the activation energy and transition state through an NEB calculation.
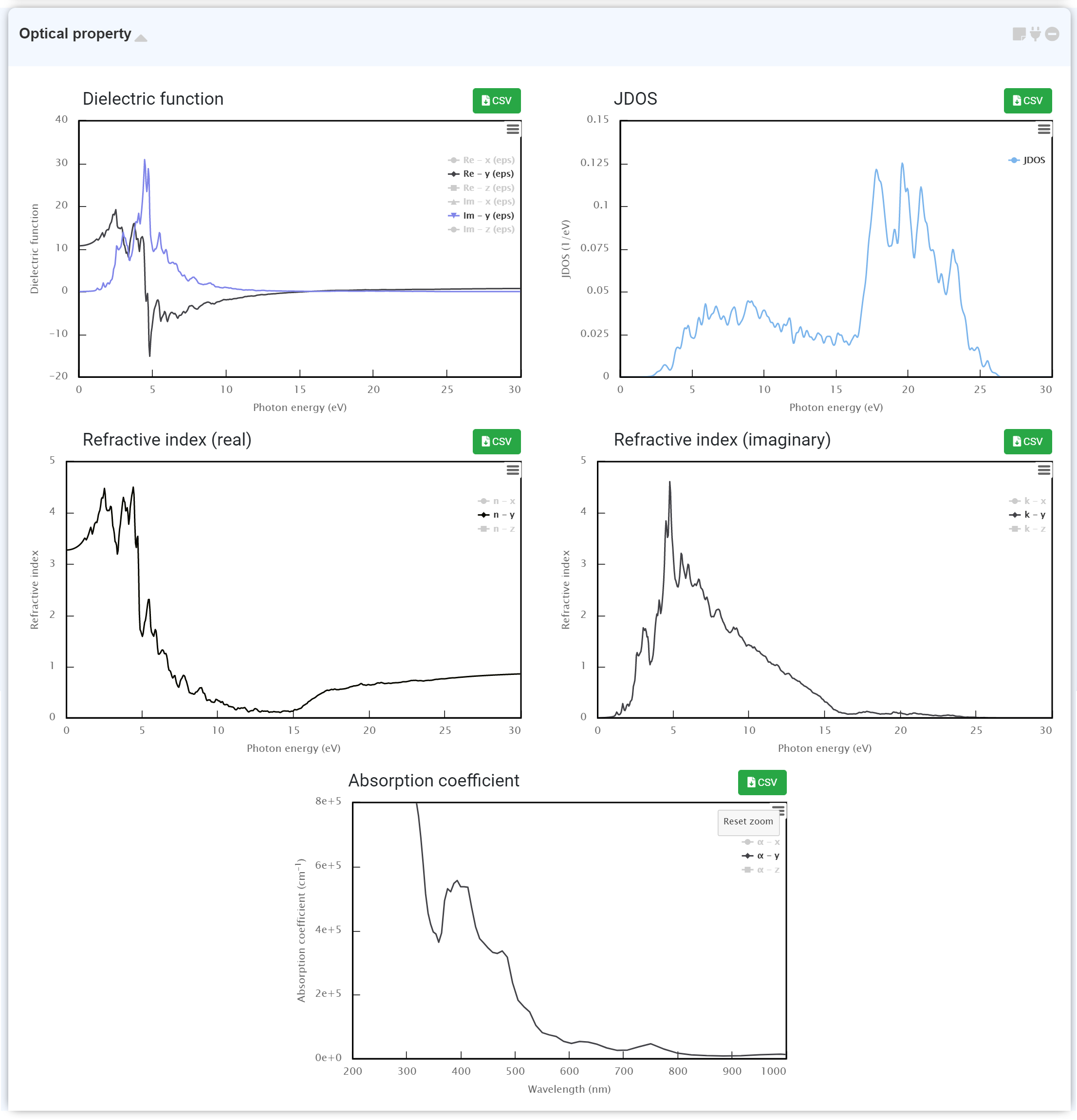
Optical property
You can obtain the optical properties that occurred from light absorption to understand the optical property in a solid.